2025 单细胞 RNA 测序数据 LLM
2025 单细胞 RNA 测序数据 LLM
2024 年单细胞大模型最近出了不少,现在来整理一下。
单细胞大模型和语言大模型类似,以 transformer 为基础的模型在大规模数据上进行训练,微调后适应各个下游任务。
scBERT as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data

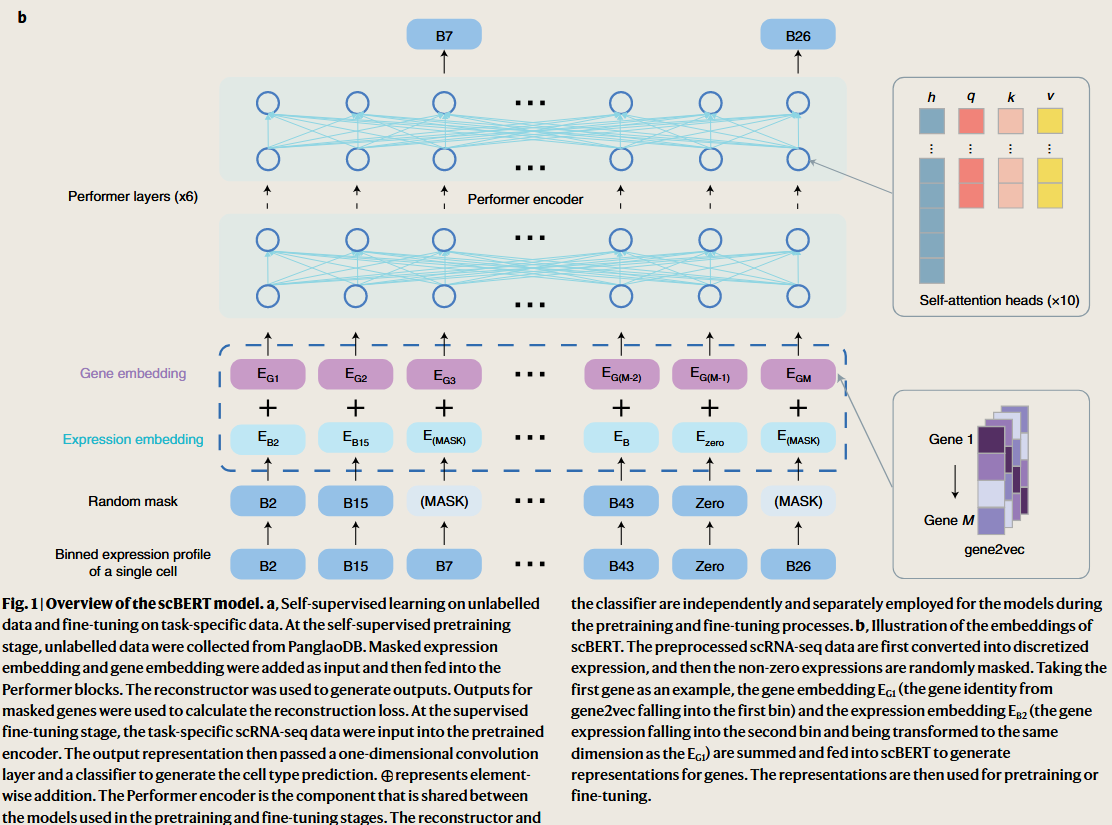
scBERT 模型采用了 BERT 的先进范式,并定制了架构来解决单细胞数据分析。scBERT 与 BERT 的联系如下。首先,scBERT 遵循 BERT 的革命性方法进行自监督预训练,并使用 Transformer 作为模型 backbone。其次,scBERT 的嵌入设计在某些方面与 BERT 相似,但具有利用基因知识的独特功能。从这个角度来看,scBERT 的 expression 嵌入可以被视为 BERT 的 token 嵌入。由于打乱输入的列并不会改变其含义(就像使用 TaBERT 扩展 BERT 来理解表格数据一样),因此绝对位置对于基因来说是没有意义的。相反,scBERT 使用 gene2vec 来生成 gene 嵌入,这可以被视为相对嵌入,它捕获任何两个基因之间的语义相似性。第三,具有全局感受野的 Transformer 可以在没有绝对位置信息的情况下有效学习全局表示和长程依赖,在非序列数据(例如图像、表格)上取得优异的性能。
Gene Embeddings
在 NLP 中,BERT 模型的输入是词嵌入,即预定义向量空间中表示各个单词的一组实值向量。词嵌入技术通过确保具有相似含义的单词具有相似的表示来帮助更好地表示文本。然而,从 scRNA-seq 的角度来看,输入是由单个基因构成的,需要预先定义的向量空间来表示它们之间的相似性。因此,scBERT 使用 gene2vec 来专门编码基因嵌入。这样,借助过去的知识提供的基因间关系,降低了模型训练的难度。
Expression Embeddings
尽管有基因嵌入,但如何利用每个基因的转录水平也存在挑战,因为每个基因的转录水平实际上是一个连续变量。值得注意的是,单词在文本中出现的频率对于文本分析来说是有价值的信息,并且通常通过词频统计分析转化为词袋,用于 NLP 领域的下游任务。基因表达也可以被认为是生物系统中已被充分记录的每个基因的出现。根据这一见解,scBERT 应用了传统使用的词频分析方法,通过分箱将连续表达变量离散化,并将其转换为 200 维向量,然后将其用作 scBERT 模型的标记嵌入。
Model building
以 Transformer 为基本单元的 BERT 模型的二次计算复杂度不能很好地扩展到长序列,而 scRNA-seq 的基因数量可达 20,000 以上。为此,采用了矩阵分解版本的 Transformer(即 Performer)来扩大序列长度。 Transformer 中的常规点积注意力是 Q、K、V 的映射,它们分别是为每个单元创建的输入查询、键和值的编码表示。双向注意力矩阵的公式为: \[ \mathrm{Att}(Q,K,V) = D^{-1}(QK^T)V, D=\mathrm{diag}(QK^T 1_{L}) \tag{1} \] 其中 \(Q = W_{Q}X\)、\(K = W_{K}X\)、\(V = W_{V}X\) 是输入 \(X\) 的线性变换; \(W_{Q}, W_{K}、W_{V}\) 为作为参数的权重矩阵; \(1_L\) 是长度为 \(L\) 的全 1 向量; \(\mathrm{diag}(\cdot)\) 是以输入向量为对角线的对角矩阵。
Performer 中的注意力矩阵描述如下:
\[ \widehat{\mathrm{Att}}(Q,K,V) = \hat{D}^{-1}(Q'((K')^T 1_{K})) \tag{2} \] 其中 \(Q'=\emptyset(Q), K'=\emptyset(K)\),函数 \(\emptyset(x)\) 定义为:
\[ \emptyset (X) = \frac{c}{\sqrt{ m }}f(\omega^TX) \tag{3} \]
其中 \(c\) 是正常数,\(\omega\) 是随机特征矩阵,\(m\) 是矩阵的维数。在这里,scBERT 构建了具有六个 Performer 编码器层和每层十个头的模型。
模型训练过程包含两个阶段:对未标记数据进行自监督学习以获得预训练模型,以及对特定细胞类型注释任务进行监督学习以获得微调模型。
Self-supervised learning on unlabelled data
在本研究中,scBERT 遵循 NLP 任务中 BERT 模型的传统自学习策略,随机屏蔽输入数据值并根据剩余输入进行预测。考虑到丢失零现象,scBERT 随机屏蔽非零基因表达,然后使用剩余基因通过模型预测重建原始输入。scBERT 利用交叉熵损失作为重建损失,公式为:
\[ L_{rec} = -\sum_{i=1}^M\sum_{j=1}^N y_{ij}\log(p_{ij}) \tag{4} \] 其中 \(M\) 是细胞的数量,\(N\) 是掩蔽基因表达值的数量; \(y_{i,j}\) 和 \(p_{i,j}\) 分别是细胞 \(i\) 中基因 \(j\) 的真实表达和预测表达。通过这种自我监督策略,该模型可以在大量未标记数据上学习基因表达模式的一般深度表示,这可能减轻下游微调过程的工作量。
Supervised learning on specific tasks
scBERT 的输出是每个基因对应的 200 维特征,并应用一维卷积对每个基因特征进行抽象信息提取。然后应用三层神经网络作为分类头,并将基因特征转换为每种细胞类型的概率。交叉熵损失也被用作细胞类型标签预测损失,计算如下:
\[ L_{Pred} = -\sum_{i=1}^{M} z_{i} \log(q_{i}) \tag{5} \]
其中 \(z_i\) 和 \(q_i\) 分别表示细胞 \(i\) 的真实细胞类型标签和预测标签。 ## scGPT: toward building a foundation model for single-cell multi-omics using generative AI
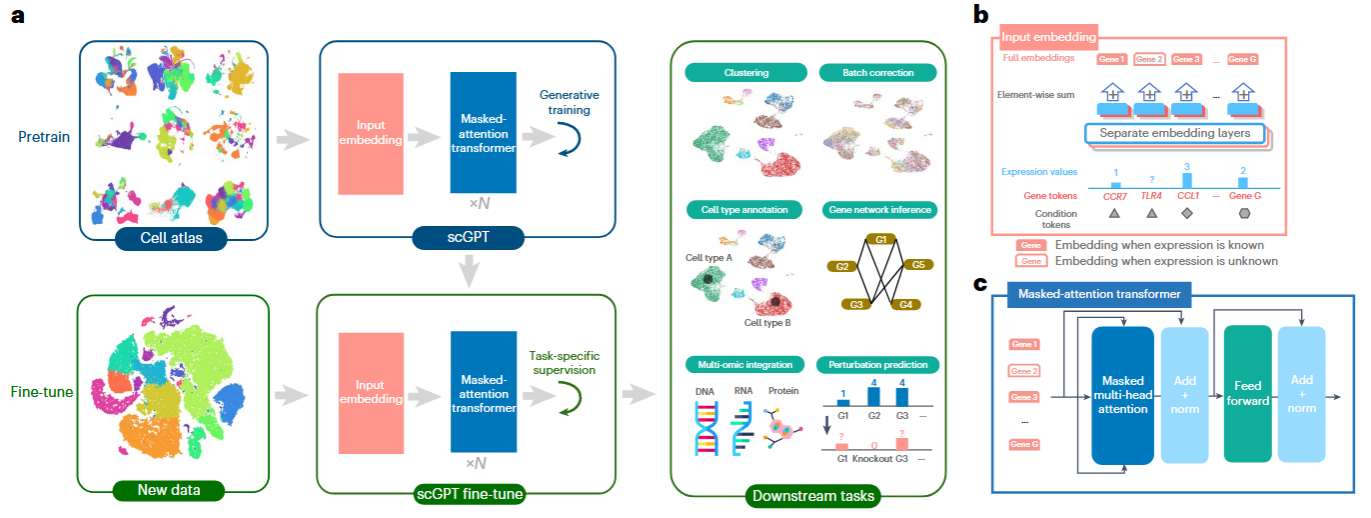
Input embeddings
单细胞测序数据被处理成逐个基因的矩阵,\(\mathbf{X} \in \mathbb{R}^{N \times G}\),其中每个元素 \(\mathbf{X}_{i,j} \in \mathbb{R}^+\) 代表 scRNA-seq 数据的 RNA 分子的读取计数或用于 scATAC-seq 数据的峰区域的染色质可及性(chromatin accessibility)。具体来说,对于 scRNA-seq 数据,该元素表示单元格 \(i \in \{0, 1, ..., N\}\) 中基因 \(j \in \{0, 1, ..., G\}\) 的 RNA 丰度。在后续部分中,scGPT 将该矩阵称为原始计数矩阵。 scGPT 的输入由三个主要部分组成:(1) 基因(或峰值)(gene (or peak))tokens、(2) 表达值和 (3) 条件(condition)tokens。对于每个建模任务,基因 tokens 和表达值均根据原始计数矩阵 \(\mathbf{X}\) 进行相应的预处理。
Gene tokens 在 scGPT 框架中,每个基因被认为是最小的信息单位,类似于 NLG 中的单词。因此,scGPT 使用基因名称作为 tokens,并为每个基因 \(g_j\) 分配一个唯一的整数标识符 \(id(g_{j})\)。这些标识符构成了 scGPT 中使用的 tokens 词汇表。这种方法提供了极大的灵活性,可以协调具有不同基因集(即由不同测序技术或预处理流程生成)的多项研究。具体来说,通过研究中所有基因的并集,可以将不同的基因 tokens 集合集成到通用词汇表中。此外,scGPT 在词汇表中加入了特殊的 tokens,例如用于将所有基因聚合到细胞表示中的 <cls> 以及用于将输入填充到固定长度的 <pad>。从概念上讲,scGPT 将 NLG 中的基因 tokens 和单词 tokens 进行比较。因此,每个单元 \(i\) 的输入基因标记由向量 \(t_g^{(i)} \in \mathbb{N}^M\) 表示:
\[ t_g^{(i)} = [id(g_1^{(i)}, id(g_2^{(i)}), ..., id(g_M^{(i)})] \tag{1} \]
Expression values 基因表达矩阵 \(\mathbf{X}\) 在用作建模输入之前需要进行额外处理。基因表达建模的一个基本挑战是不同测序方案的绝对量值的变化。测序深度的变化和稀疏表达基因的存在导致不同批次的测序样本之间的数据规模存在显着差异。这些差异很难通过常见的预处理技术(例如每百万转录本归一化和 log1p 转换,transcripts-per-million normalization and log1p transformation)来缓解。即使在这些转换之后,相同的绝对值也可以在不同的测序批次中传达不同的“语义”含义。为了解决这种规模差异,scGPT 提出了值分箱(value binning)技术,将所有表达计数转换为相对值。对于每个细胞中的每个非零表达计数,scGPT 计算原始绝对值并将它们划分为 \(B\) 个连续区间 \([b_{k}, b_{k+1}]\),其中 \(k \in \{1, 2, ..., B\}\)。每个区间代表所有表达基因的相等部分 (\(1/B\))。值得注意的是,为每个细胞计算一组新的 bin 边缘,因此间隔边缘 \(b_{k}\) 可能因细胞而异。细胞 \(i\) 的分箱表达式值 \(x^{(i)}_{j}\) 定义为:
\[ x_{j}^{(i)} = \begin{cases} k, & if\ \mathbf{X}_{i,j}>0\ and \ \mathbf{X}_{i,j} \in [b_{k}, b_{k+1}] \\ 0, & if\ \mathbf{X}_{i,j}=0. \end{cases} \tag{2} \]
通过这种分箱技术,\(x^{(i)}_{j}\) 的语义在不同测序批次的细胞之间是一致的。例如,\(x^{(i)}_j = B\) 的值始终表示基因中的最高表达。值得注意的是,对于微调任务,scGPT 还在值装箱步骤之前执行了 log1p 转换和 HVG 选择。为了简化符号,scGPT 使用 \(\mathbf{X}_{i,j}\) 来表示分箱之前的原始数据矩阵和预处理后的数据矩阵。因此,细胞 \(i\) 的分箱表达值的最终输入向量表示为
\[ x^{(i)} = [x_{1}^{(i)}, x_{2}^{(i)}, \cdots, x_{M}^{(i)}] \tag{3} \]
Condition tokens 条件 tokens 包含与单个基因相关的各种元信息,例如微扰实验改变(由微扰 tokens 表示)。为了表示位置条件 tokens,scGPT 使用与输入基因共享相同维度的输入向量。该向量表示为:
\[ t_{c}^{(i)} = [t_{c,1}^{(i)}, t_{c,2}^{(i)}, \cdots, t_{c,M}^{(i)}], \tag{4} \]
其中 \(t^{(i)}_{c,j}\) 表示条件对应的整数索引。
Embedding layers scGPT 使用传统的嵌入层(即PyTorch嵌入层(https://pytorch.org/docs/stable/ generated/torch.nn.Embedding.html))\(\mathrm{emb}_{g}\) 和 \(\mathrm{emb}_{c}\) 分别作为基因 tokens 和条件 tokens,以便于将每个 tokens 映射到维度为 \(D\) 的固定长度嵌入向量。scGPT 使用全连接层(表示为 \(\mathrm{emb}_{x}\) )作为分箱表达值来增强表达能力。这种选择使得能够对基因表达值的顺序关系进行建模。因此,细胞 \(i\) 的最终嵌入 \(\boldsymbol{h}^{(i)} \in \mathbb{R}^{R \times D}\) 定义为:
\[ \boldsymbol{h}^{(i)} = \mathrm{emb}_{g}(\boldsymbol{t}_{g}^{(i)}) + \mathrm{emb}_{x}(\boldsymbol{x}^{(i)}) + \mathrm{emb}_{c}(\boldsymbol{t}_{c}^{(i)}) \tag{5} \]
Cell and gene expression modeling by transformers
scGPT transformer scGPT 使用自注意力 transformer 对公式(5)中的完整输入嵌入 \(\boldsymbol{h}^{(i)}\) 进行编码。自注意力机制对 \(M\) 个嵌入向量的序列进行操作,使其特别适合捕获基因之间的相互作用。堆叠 transformer 块的输出可以定义如下:
\[ \begin{align} \boldsymbol{h}^{(i)}_{0} & = \boldsymbol{h}^{(i)} \\ \boldsymbol{h}^{(i)}_{l} & = \mathrm{transformer\_block}(\boldsymbol{h}_{l-1}^{(i)}) \ \forall l \in [1,n] \end{align} \tag{6} \]
scGPT 将得到的表示 \(\boldsymbol{h}_{n}^{(i)}\) 用于基因级和细胞级任务。直接应用基因级微调目标(Fine-tuning Objectives)。示例包括基因表达预测 (GEP,gene expression prediction) 目标和扰动表达预测任务 (perturb-GEP,perturbed expression-prediction task)。对于细胞级任务,scGPT 首先将 \(\boldsymbol{h}_{n}^{(i)}\) 集成到细胞嵌入向量(细胞表示)中。一个例子是细胞类型分配任务,其中细胞嵌入用于通过细胞类型分类训练目标中添加的分类器来预测细胞类型标签。
输入维度 \(M\) 可以达到数万个基因,大大超过了 NLG 中常用的传统 transformer 的输入长度。为了应对这一挑战并确保有效的自注意力机制,scGPT 利用 FlashAttention 的加速自注意力实现。该实现有效增强了模型容量,并能够有效处理大输入维度。尽管采用了 FlashAttention,但任何高效的 Transformer 也都可以用于 scGPT,例如具有线性复杂度的 Transformer (Linformer) 和 Kernelized Self-Attention (KSA)。
Cell representation 每个细胞类似于一个由基因组成的“句子”,其表示 \(\boldsymbol{h}_{c}^{(i)} \in \mathbb{R}^D\) 是通过聚合学习到的基因级表示 \(\boldsymbol{h}_{n}^{(i)}\) 获得的。在这种情况下可以很容易地使用各种池化操作,例如逐元素平均池化或加权池化。在本研究中,scGPT 选择使用特殊的 token <cls> 来表示细胞,使模型能够学习 transformer 块内的池化操作。 <cls> token 被附加到输入 tokens 的开头,并且该位置的最终嵌入被提取为细胞表示。因此,单元嵌入 \(\boldsymbol{h}_{c}^{(i)}\) 可以通过堆叠的最终层嵌入 \(\boldsymbol{h}_{n}^{(i)}\) [<cls>] 中的相应行来提取,其中 [<cls>] 操作检索 < cls > token 位置索引处的行。
Representation for batch and modality scGPT 使用额外的 tokens 集来表示不同的测序批次和测序模式(来自 RNA-seq 的基因、来自 ATAC-seq 的峰值等),专门用于 scRNA-seq 和 scMultiomic 集成任务。这类似于输入嵌入中引入的条件 tokens,并且使用标准嵌入层类似地实现。模态 tokens \(t_{m}^{(i)}\) 与各个输入特征 \(g_j\) 相关联(例如,指示它是否是基因、区域或蛋白质)。批次 tokens 最初位于细胞级别,但也可以传播到单个细胞的所有特征。换句话说,相同的批量 tokens \(t_{b}^{(i)}\) 可以重复单个细胞 \(i\) 的输入特征的至长度 \(M\):
\[ t_{b}^{(i)} = [t_{b,1}^{(i)}, t_{b,2}^{(i)}, \dots, t_{b,M}^{(i)}] = [t_{b}^{(i)}, t_{b}^{(i)}, \dots, t_{b}^{(i)}] \]
输入嵌入中描述的 tokens 与批处理和模态 tokens 之间的区别在于,批处理和模态 tokens 的这些嵌入不用作 transformer 块的输入。相反,它们在进入特定的微调目标之前与特征或细胞级别的 transformer 输出连接。这是为了防止 transformer 放大相同模态特征的注意力,同时低估不同模态的特征。此外,了解模态和/或批次身份有助于下游微调目标中的基因表达建模。当模型学习预测以模态和/或批次标识符为条件的表达值时,这种偏差会从基因和细胞表示本身中隐式消除。这是一种促进批量校正的技术。
例如,在 scMultiomic 集成任务中,scGPT 将 transformer 输出与批次和模态嵌入的总和连接起来。这作为表达建模下游微调目标的输入:
\[ \boldsymbol{h}_{n}^{'(i)} = \mathrm{concat}(\boldsymbol{h}_{n}^{(i)}, \mathrm{emb}_{b}(\boldsymbol{t}_{b}^{(i)}) + \mathrm{emb}_{m}(\boldsymbol{t}_{m}^{(i)})), \tag{8} \] 其中 \(\mathrm{emb}_{b}\) 和 \(\mathrm{emb}_{m}\) 分别表示批处理嵌入层和模态嵌入层。 \(\boldsymbol{h}_{n}^{(i)}\) 表示 transformer 层(scGPT transformer)的输出。
或者,在 scRNA-seq 集成任务中,批量嵌入与细胞表示的串联会产生以下表示作为输入:
\[ \boldsymbol{h}_{c}^{'(i)} = \mathrm{concat}(\boldsymbol{h}_{c}^{(i)}, \mathrm{emb}_{b}(\boldsymbol{t}_{b}^{(i)})), \tag{9} \]
其中 \(t_{b}^{(i)}\) 表示细胞 \(i\) 的批次标识。 \(\boldsymbol{h}_{c}^{(i)}\) 是微调目标中的原始细胞表示。请注意,修改后的版本 \(\boldsymbol{h}_{c}^{'(i)}\) 仅与表达建模目标相关,不适用于基于分类的目标,如微调目标中详述。
Generative pretraining
Foundation model pretraining 基础模型被设计为一个通用的特征提取器,可以使各种下游任务受益。预训练中使用的 tokens 词汇包含人类基因组中的整套基因。在模型预训练(输入嵌入)之前对表达式值进行分箱。为了加快训练速度,scGPT 将输入限制为每个输入单元仅具有非零表达的基因。为了有效地训练模型以捕获基因-基因关系和基因-细胞关系,scGPT 引入了一种具有专门注意掩模的生成训练策略,如下一节所述。
Attention mask for generative pretraining 自注意力已被广泛用于捕获 tokens 之间的共现模式。在自然语言处理中,这主要通过两种方式实现:(1)BERT 和 RoBERTa 等 Transformer 编码器模型中使用的屏蔽 tokens 预测,其中输入序列中的随机屏蔽 tokens 在模型的输出中进行预测;(2)在因果 Transformer 解码器模型(例如 OpenAI GPT 系列)中进行顺序预测的自回归生成。 OpenAI GPT-3(参考文献 19)和 GPT-4(参考文献 20)中使用的生成预训练使用统一的框架,其中模型根据由已知输入 tokens 组成的“prompt”来预测最有可能的下一个 token。该框架提供了极大的灵活性,可用于各种 NLG 应用程序,并展示了零样本和微调设置中的上下文感知等功能。scGPT 相信,生成训练可以以类似的方式有益于单细胞模型。具体来说,scGPT 对两个任务感兴趣:(1)根据已知基因表达生成未知基因表达值,即通过“gene prompts”生成,以及(2)给定输入细胞类型条件生成全基因组表达,即即,通过“cell prompts”生成。
尽管 tokens 和 prompts 的想法相似,但由于数据的非顺序性质,对基因读取进行建模本质上不同于自然语言。与句子中的单词不同,细胞内基因的顺序是可以互换的,并且没有可预测的“下一个基因”的等效概念。这使得将 GPT 模型中的因果掩蔽公式直接应用于单细胞数据变得具有挑战性。为了应对这一挑战,为 scGPT 开发了一种专门的注意力屏蔽机制,该机制根据注意力分数定义预测顺序。
注意掩蔽通常可以应用于 Transformer 块中的自注意力图:对于 \(M\) 个基因 tokens(公式(1))的输入,第 (\(l + 1\)) 个 Transformer 块将多头自注意力应用于其 M 个 tokens 的输入 \(\boldsymbol{h}_{l}^{(i)} \in \mathbb{R}^{M \times D}\)(公式(6))。具体来说,每个自注意力操作的计算如下:
\[ \begin{align} &Q = \boldsymbol{h}_{l}^{(i)}W_{q}, K = \boldsymbol{h}_{k}^{(i)}W_{k}, V =\boldsymbol{h}_{k}^{(i)}W_{v}, \\ &\mathrm{Attention}(Q,K,V) = \mathrm{softmax} \left( \frac{QK^T}{\sqrt{ d }} + \mathbf{A}_{mask}\right)V \end{align} \tag{10} \]
其中 \(Q, K, V \in \mathbb{R}^{M \times d}\) 表示查询、键和值向量。 \(W_{q}、W_{k}、W_{v} \in \mathbb{R}^{D \times d}\) 是可学习的权重矩阵。 \(d\) 是特征维度,用作 \(\frac{QK^T}{\sqrt{ d }}\) 中的缩放因子以保持数值稳定性。注意力掩码 \(\mathbf{A}_{mask} \in \{ 0,-\mathrm{inf} \}^{M\times M}\) 通过修改查询和键之间的原始注意力权重来描绘自注意力的范围,如 \(\frac{QK^T}{\sqrt{ d }}\) 中所示。具体来说,将 \(-\mathrm{inf}\) 添加到矩阵中的位置 (\(i, j\)) 会使 softmax 之后的注意力权重无效,从而禁止第 \(i\) 个查询和第 \(j\) 个键之间的注意力。另一方面,添加 0 意味着注意力权重保持不变。这种注意力屏蔽技术允许模型专注于特定的上下文元素。
专门设计了 scGPT 注意力掩码,以统一的方式支持 gene-prompt 和 cell-prompt 生成。注意掩码 \(\mathbf{A}_{mask} \in \{ 0,-\mathrm{inf} \}^{M\times M}\) 在补充图 1a 中可视化,其中查询按行组织,键按列组织。如图底部注释,输入嵌入 \(\boldsymbol{h}_{l}^{(i)}\) 中的每个 token 可以是以下三组之一:(1) 用于细胞嵌入的保留 <cls> 标记(在细胞表示中引入),(2)具有 token 嵌入和表达值嵌入的已知基因和(3)要预测表达值的未知基因。 scGPT 注意力屏蔽的经验法则是只允许在“已知基因”的嵌入和查询基因本身之间进行注意力计算。这是通过使用 \(\mathbf{A}_{mask}\) 中的元素 \(a_{i,j}\) 来实现的,如下所示:
\[ a_{i,j} = \begin{cases} 0, & if\ j\ \not\in unknown\ genes, \\ 0, & if\ i = j\ and\ j \in unknown genes, \\ -\mathrm{inf}, & if\ i \not= j\ and\ j\ \not\in unknown\ genes. \end{cases} \]
在每一代迭代中,scGPT 预测一组新基因的基因表达值,这些基因又成为下一次迭代中的“已知基因”,用于注意力计算。这种方法通过在非顺序单单元数据中进行顺序预测,反映了传统 transformer 解码器中下一个 token 预测的因果屏蔽设计。
如补充图 1a 所示,在训练过程中,scGPT 随机选择一部分基因作为未知基因,以便在输入中省略它们的表达值。注意力仅应用于已知基因和查询未知基因本身之间,而不应用于其他未知基因的位置。例如,要在位置 \(j\) 预测的基因具有细胞嵌入、已知基因及其自身的注意力分数,但没有其他未知基因,如注意力掩模的最后一行所示。 scGPT 模型通过堆叠的 Transformer 块和上述的屏蔽注意力图来预测这些未知基因的表达。推理步骤如补充图 1b 所示。在细胞提示生成的推理过程中,scGPT 会生成以特定细胞类型为条件的所有全基因组基因表达。在表示细胞类型条件的第一位置处输入经过训练的细胞嵌入。数千个基因表达值的整个生成过程是在 \(K\) 个迭代步骤中进行的(即补充图 1b 中的 \(K = 3\) 个步骤)。例如,在一次迭代中 \(i \in \{ 1,2,\dots,K \}\),注意力掩蔽机制允许关注之前 0 到 \(i − 1\) 次迭代中的所有预测基因。在每次迭代中,scGPT 从未知集中选择预测置信度最高的前 \(1/K\) 基因,作为下一次迭代 \(i + 1\) 中的已知基因。直观地说,该工作流程以自回归方式简化了基因表达的生成,其中首先生成具有最高预测置信度的基因表达值并用于帮助后续几轮生成。基因提示生成以迭代方式类似地工作。不同之处在于,它从一组具有观察到的表达值的已知基因开始,而不是细胞嵌入。
scGPT 注意力掩蔽统一了已知基因的编码过程和未知基因的生成。它也是第一个对非序列数据进行自回归生成的 transformer 方案之一。
Learning objective for pretraining scGPT 使用基因表达预测目标来优化模型以预测未知基因的表达值。具体来说,scGPT 使用多层感知器网络(MLP)来估计未知表达值并计算均方损失 \(\mathcal{L}\):
\[ \mathcal{L} = \frac{1}{|\mathcal{U}_{unk}|} \sum_{j \in \mathcal{U}_{unk}} (\mathrm{MLP}(\boldsymbol{h}_{n}^{(i)}) - x_{j}^{(i)})^2, \tag{12} \]
其中 \(\mathcal{U}_{unk}\) 表示未知基因的输出位置集合,\(x_{j}^{(i)}\) 是要预测的实际基因表达值。 \(|\cdot|\) 运算检索集合的元素数量。
正如生成预训练的注意力掩码中提到的,支持 gene-prompt 和 cell-prompt 生成。训练时,这两种模式是连续进行的。在一个给定细胞的输入基因 tokens 中,选择一部分基因作为“未知”基因,并省略它们的表达值。首先,在 gene-prompt 步骤中,模型的输入包含 <cls> token 嵌入、已知基因嵌入和未知基因嵌入。损失(公式(12))是使用模型的输出计算的。其次,在 cell-prompt 步骤中,使用上一步的输出 cell embedding(即细胞表示中的 \(\boldsymbol{h}_{c}^{(i)}\))来替换 <cls> 位置处的 embedding。其他计算保持不变。最后,两者的损失值步骤被加在一起并用于计算梯度以优化模型参数。
Fine-tuning objectives
scGPT 利用各种微调目标来促进细胞和基因的生物学有效表示的学习,以及用于批量校正等正则化目的。
Gene expression prediction 为了鼓励学习基因-基因相互作用,scGPT 结合了 GEP。此微调目标的工作原理与预训练中的目标(预训练的学习目标)类似,但适用于屏蔽位置。具体来说,对于每个输入细胞,基因 tokens 的子集及其相应的表达值 \(\boldsymbol{x}^{(i)}\) 被随机屏蔽。 scGPT 经过优化,可以准确预测屏蔽位置的表达值。这种微调目标有利于模型有效地编码数据集中基因之间的共表达。目标最小化掩蔽位置处的均方误差,表示为 \(\mathcal{M}_{\mathrm{mask}}\)。 GEP 的工作原理如下:
\[ \begin{align} \tilde{\boldsymbol{x}}^{(i)} & = \mathrm{MLP}(\boldsymbol{h}_{n}^{(i)}), \\ \mathcal{L}_{\mathrm{GEP}} & = \frac{1}{|\mathcal{M}_{\mathrm{mask}}|} \sum_{j \in \mathcal{M}_{\mathrm{mask}}} (\tilde{x}_{j}^{(i)} - x_{j}^{(i)}). \end{align} \tag{13} \]
这里, \(\tilde{\boldsymbol{x}}^{(i)} \in \mathbb{N}^M\) 表示细胞 \(i\) 的表达估计的行。值得注意的是,如果提供了测序批次或模态条件,scGPT 使用公式(8)中的 \(\boldsymbol{h}_{n}^{'(i)}\) 而不是 \(\boldsymbol{h}_{n}^{(i)}\)。
GEP 提出了一个通用的自我监督微调目标,旨在预测基因表达值。在某些下游任务中,例如扰动预测,模型需要预测扰动的基因表达值而不是原始值。scGPT 将这种变化称为扰动-GEP。scGPT 维持公式(13)中的 MLP 估计量,但使用扰动后基因表达作为目标 \(x_{j}^{(i)}\)。在扰动-GEP 中,模型应该预测所有输入基因的扰动后表达。
Gene expression prediction for cell modeling 此微调目标的操作与 GEP 类似,但根据细胞表示 \(\boldsymbol{h}_{c}^{(i)}\) 预测基因表达值,以明确促进细胞表示学习。对于输入细胞 \(i\) 中的每个基因 \(j\),scGPT 创建一个查询向量 \(q_j\) 并使用 \(q_j\) 和细胞表示 \(\boldsymbol{h}_{c}^{(i)}\) 的参数化内积作为预测表达值:
\[ \begin{align} \boldsymbol{q}_{j} & = \mathrm{MLP}(\mathrm{emb}_{g}(\boldsymbol{t}_{g}^{(i)})), \\ \tilde{x}_{j}^{(i)} & = \boldsymbol{q}_{j} \cdot \boldsymbol{W}\boldsymbol{h}_{c}^{(i)}, \\ \mathcal{L}_{\mathrm{GEPC}} & = \frac{1}{|\mathcal{M}_{\mathrm{mask}}|} \sum_{j \in \mathcal{M}_{\mathrm{mask}}} (\tilde{x}_{j}^{(i)} - x_{j}^{(i)}). \end{align} \tag{14} \] 用于细胞建模的 GEP (GEPC) 继承了公式 (5) 中的基因 tokens 嵌入 \(\mathrm{emb}_{g}(\boldsymbol{t}^{(i)}g)\)。在集成任务中,scGPT 使用公式(9)中的 \(\boldsymbol{h}_{c}^{'(i)}\) 代替 \(\boldsymbol{h}_{c}^{(i)}\)。在 scGPT 的实验中,scGPT 观察到,与单独使用任一方法相比,结合使用 GEP 和 GEPC 可以显着提高性能。
Elastic cell similarity 这种微调目标通过利用相似性学习损失来增强细胞表示
\[ \mathcal{L} = - (\mathrm{sim} (\boldsymbol{h}_{c}^{(i)}, \boldsymbol{h}_{c}^{(i')}) - \beta), \tag{15} \]
其中 sim 表示余弦相似度函数,而 \(i\) 和 \(i'\) 表示小批量中的两个细胞。另外,\(\beta\) 表示预定义的阈值,ECS 是弹性细胞相似度。这种方法背后的基本思想是增强余弦相似度值高于 \(\beta\) 的对之间的相似度,从而使它们更加相似。相反,鼓励不同的配对保持更远的距离。
Domain adaptation via reverse back propagation 细胞表征学习因批次效应的存在而受到阻碍,批次效应是由测序技术引入的非生物批次差异造成的。为了缓解这个问题,scGPT 使用不同的 MLP 分类器根据细胞表示 \(\boldsymbol{h}_{c}^{(i)}\) 来预测与每个输入细胞相关的测序批次,并通过反转模型内的梯度来修改反向传播过程。这种方法利用了 Ganin 和 Lempitsky 提出的稳健域适应方法的见解。
Cell type classification 这个微调目标旨在利用学习的细胞表示来注释单个细胞。使用单独的 MLP 分类器根据细胞表示 \(\boldsymbol{h}_{c}^{(i)}\) 来预测细胞类型。该微调目标通过预测细胞类型概率和真实标签之间的交叉熵损失进行优化。
Fine-tuning on downstream tasks
Cell type annotation 对于细胞类型注释任务,scGPT 在带有真实标签的参考集上对模型进行了微调,并在保留的查询集上验证了注释性能。保留预训练基础模型和参考集之间的通用基因标记集。在模型微调之前,对基因表达值进行归一化、对数转换和分箱。除了随机初始化的输出细胞类型分类器之外,所有预训练的模型权重都用于初始化微调模型。所有具有零和非零表达值的基因标记都用于训练。细胞类型分类微调目标用于最小化分类损失。
Perturbation response prediction 为了微调扰动预测任务,scGPT 在模型训练之前选择了 HVG 并预处理了表达式值。预训练模型中的嵌入层和 tranformer 层的参数用于初始化微调模型。在微调过程中,所有具有零和非零表达值的基因标记都被包括在内。扰动预测任务中的输入采用了两个显着的变化:首先,scGPT 使用 log1p 转换的表达值作为输入和目标值,而不是分箱值,以更好地预测该任务的绝对扰动后表达。其次,scGPT 在每个输入基因位置附加一个二元条件 token,以指示该基因是否受到干扰。scGPT 采用了扰动 GEP 微调目标,并对训练设置进行了进一步修改。scGPT 没有使用同一细胞的屏蔽和未屏蔽表达值作为输入和学习目标,而是使用控制细胞作为输入,扰动细胞作为目标。这是通过将未受扰动的控制单元与每个受扰动的单元随机配对以构建输入-目标对来实现的。输入值由对照细胞中的所有基因表达值组成。因此,该模型学会了根据控制基因表达和扰动标记来预测扰动后的响应。
Perturbation response prediction 当输入原始计数矩阵包含来自不同测序批次或技术的多个数据集时,批次效应可能是细胞类型聚类中的主要混杂因素。因此,scGPT 的目标是在整合多个 scRNA-seq 数据集时纠正批次效应,同时保留生物方差。为了对此集成任务进行微调,保留了预训练基础模型和当前数据集之间的通用基因标记集。scGPT 进一步从公共集中选择了 HVG 的子集作为输入。scGPT 在模型训练之前对表达值进行预处理,类似于细胞类型注释任务。所有预训练的模型权重都用于初始化微调模型。默认情况下,所有具有零和非零表达值的基因标记都用于训练。除了 GEP 和 GEPC 之外,还同时优化了 ECS、通过反向反向传播 (DAR) 进行的域适应和 DSBN 微调目标,以通过反向反向传播和特定域归一化来增强单元对比学习和显式批量校正。
Integrative representation learning for scMultiomic data scMultiomic 数据可能包含跨实验批次的不同测序模式。scGPT 检查了 scMultiomic 数据的两种数据集成设置(配对和镶嵌)。在配对设置中,所有样本(细胞)共享所有已测序的数据模式。在马赛克设置中,某些批次共享一些常见的数据模式,但不是全部。由于存在额外的 ATAC 和/或蛋白质标记,scGPT 继承了仅用于 RNA 数据的经过训练的基因嵌入,并从头开始训练了额外的标记嵌入和模型的其余部分。如果数据集包含额外的蛋白质数据,则仅在训练中使用具有非零表达值的标记。否则,默认情况下使用零和非零表达式值。scGPT 使用了一组额外的模态标记来指示每个标记的数据类型(即基因、区域或蛋白质),并促进 GEP 和 GEPC 微调目标中的掩蔽基因和值预测(批次和模态的表示)。默认情况下,该模型使用 GEP 和 GEPC 微调目标进行优化。如果存在多个批次,则包含 DAR 以促进多模式批次校正。
Gene regulatory network inference 对于图 5 中基于基因嵌入的 GRN 推理,在零样本设置中,scGPT 基于 k-近邻从 scGPT 的预训练基因嵌入构建了基因相似性网络。在微调设置中,scGPT 以类似的方式从在免疫人类数据集上微调的 scGPT 模型构建了基因相似性网络。继 Ceglia 等人之后,scGPT 进一步对相似度图进行 Leiden 聚类,并从由 5 个或更多基因组成的基因簇中提取基因程序。
对于图 6 中基于注意力的目标基因选择,scGPT 在 Adamson 扰动数据集上微调了 scGPT 血液模型,该数据集由针对白血病细胞系的 87 个 CRISPR 干扰实验组成。scGPT 在图 6a 中说明了目标基因选择流程。对于每个感兴趣的扰动基因,scGPT 首先通过分别向模型提供扰动细胞组和对照细胞组来检索两组注意图(扰动细胞和控制细胞)。请注意,原始注意力分数是从模型最后一个注意力层的所有八个注意力头获得的。然后,原始注意力分数进行两轮排名归一化,首先按行,然后按列。然后,对八个注意力头的排序归一化注意力分数进行平均,以输出聚合注意力图。这就得出了用于影响最大的基因选择的最终注意力图。对于每个感兴趣的扰动基因,scGPT 通过对扰动基因列中的最终注意力图的分数进行排名来选择其影响最大的基因。这反映了注意力图中的列表明感兴趣的基因对其他基因的影响程度的直觉。scGPT 提供了三种影响最大的基因选择设置,即来自控制注意力图的“控制”、来自扰动注意力图的“扰动”以及来自两者之间差异的“差异”。从控制注意图中选择的基因目标应反映感兴趣基因参与的基础途径,而扰动注意图则反映扰动后效应。这两个注意力图之间的差异应该突出显示从扰动之前到扰动之后基因网络中变化最大的边缘。
同样,对于涉及多个转录因子的基于注意力的扩展基因相互作用预测(补充说明 7),scGPT 在 Replogle 数据子集上微调了 scGPT 血液模型 34,并报告了“扰动”设置中受影响最大的基因。
Large-scale foundation model on single-cell transcriptomics
Methods
Pretraining data collection and preprocessing
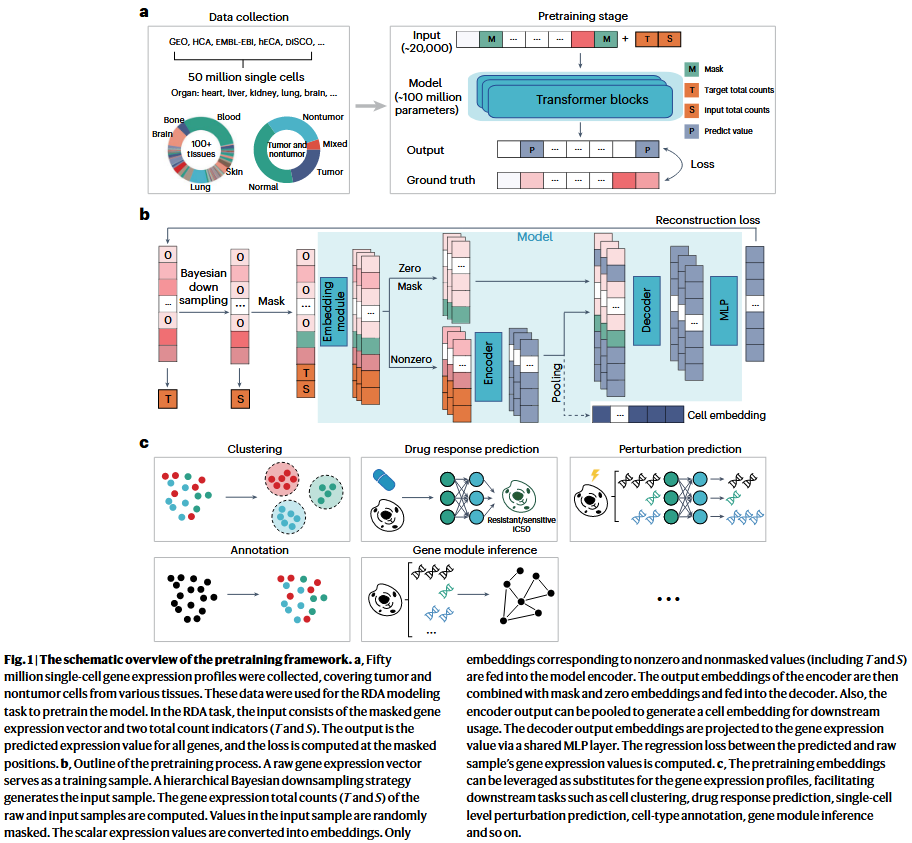
Data collection 许多人类 scRNA-seq 数据存放在 Gene Expression Omnibus (GEO)存储库、HCA、Single Cell Portal、EMBL-EBI 等中。还有一些研究整合来自多种来源的人类单细胞,例如 hECA4、DISCO7 等。这些数据库中的每个数据集都链接到已发表的研究,因此具有相应的 DOI ID。从这些数据库中手动收集 scRNA-seq 数据,并删除具有重复 ID 的数据集。大多数数据集提供了原始计数矩阵。对于具有归一化表达谱的数据集,将它们转换回原始计数形式:将原始矩阵中的最小非零值视为原始计数值 1,所有剩余的非零值除以该最小值和整数部分被带走了。对于无法转换回原始计数的每百万转录本 (TPM) 或每百万片段每千碱基转录本片段映射 (FKPM) 表达谱的数据集,保持它们不变。
scFoundation 的数据收集包括来自健康捐赠者以及各种疾病和癌症类型样本的不同器官和组织的超过 5000 万个单细胞,代表了人类单细胞转录组的全谱。将所有数据分为训练数据集和验证数据集。验证数据集是随机采样的,包含 100,000 个单细胞,并且对于所有测试模型都保持一致。
Gene symbol unification scFoundation 使用 HUGO 基因命名委员会提供的基因符号映射参考统一了所有原始计数基因表达矩阵的基因符号。 scFoundation 纳入了人类蛋白质编码基因和常见线粒体基因,总共构成了 19,264 个基因。如果某些符号丢失,会用零值填充它们。
Quality control 为了过滤受污染的空滴、极低质量的细胞和受损的细胞,使用 Seurat69 和 Scanpy61 软件包保留表达超过 200 个基因的细胞(即非零值计数 >200 的表达载体)进行预训练。
scFoundation model architecture
开发了 xTrimoGene 模型作为 scFoundation 的骨干模型。它具有三个模块:嵌入模块将标量值转换为 transformer 块所需的嵌入;编码器以非零和非掩码表达基因作为输入,使用原始 transformer 块并且具有较大的参数大小;解码器将所有基因作为输入,使用 performer 块并且具有相对较小的参数大小。消融实验表明,与其他架构相比,这种不对称设计减少了计算和内存挑战(补充表 6)。
Embedding module 给定一个细胞的基因表达值向量 \(\mathbf{X}_{\mathrm{Input}} \in \mathbb{R}^{n=19,264}\),基因 \(i\) 的表达值 \(x_{i}^{\mathrm{Input}}\) 是大于或等于 0 的连续标量。与以前的语言或最近开发的基于单细胞 transformer 的模型不同,对于每个基因 \(i\),嵌入模块直接将表达标量转换为可学习的值嵌入 \(\mathbf{X}_{i}\),而无需任何离散化。然后,将值嵌入与基因名称嵌入 \(\mathbf{T}_{i}^G\) 添加以形成最终的输入嵌入 \(\mathbf{E}_{i}^{\mathrm{ Input }}\)。值嵌入是一组嵌入的加权汇总,其中权重是从基因表达标量值中学习的。基因名称嵌入是从查找表中检索的,表中的嵌入是随机初始化的,并且可以在预训练期间学习(补充说明 8)。连续嵌入方案的消融表明,与其他值离散化方法相比, scFoundation 的设计具有优势(补充图 14)。
Encoder 编码器仅处理非零和非掩码值(即表达的基因和两个总计数)的嵌入,因此编码器的输入长度约为全基因长度的 10%。将 \(S^E = \{ S_{0}^E,S_{1}^{E},\dots,S_{K}^{E} \}\) 表示为具有 \(K\) 个元素的非零和非掩码值的索引集,编码器的输入定义为 \[ X^{\mathrm{Enc-Input}} = [\mathbf{E}_{S_{0}}^{\mathrm{Input}}, \mathbf{E}_{S_{1}}^{\mathrm{Input}}]. \] 编码器的设计大大减少了所需的计算资源,使得编码器可以采用一系列普通 transformer 块来捕获基因依赖性,而无需任何内核或低秩近似。编码器的输出是中间嵌入 \(X^{\mathrm{Inter}}\):
\[ X^{\mathrm{Inter}} = Trm(X^{\mathrm{Enc-Input}}) \in \mathbb{R}^{K \times d}, \]
其中 Trm 表示一系列 transformer 块,这些块中的核心功能是注意力机制,可以表示为 \[ \mathrm{Att} = D^{-1}AVA = \exp\left( \frac{QK^T}{\sqrt{ d }} \right), D = \mathrm{diag}(A \mathbf{1_{K}}) \] 其中 \(Q=XW_{q},K=XW_{k}\) 和 \(V=XW_{v}\) 是输入 \(X\) 的线性变换,\(W\) 是训练参数。 \(\mathbf{1_{k}}\) 是长度为 \(K\) 的全 1 向量,\(\mathrm{diag}(\cdot)\) 是以输入向量为对角线的对角矩阵。
中间嵌入 \(X^{\mathrm{Inter}}\) 有两种用途:(1)它们被发送到具有零嵌入和掩码嵌入的解码器中,(2)它们被汇集为细胞嵌入以供下游使用。
Decoder 为了建立全转录组基因调控关系,还应考虑使用零表达基因来恢复掩模位置的表达值。来自编码器的中间嵌入与零嵌入和掩码嵌入连接起来,形成具有完整基因长度的解码器输入张量 \(X^{\mathrm{Dec-Input}}\)
其中 \(K_{0}\) 和 \(K_{m}\) 分别是零嵌入和屏蔽嵌入的数量。使用基于内核的近似变换器变体 Performer 作为解码器的骨干,因为注意力计算对于长序列具有挑战性。在 Performer 中,使用了可内核化的注意力机制:
\[ \overline{Att}(Q,K,V)=\hat{D}^{-1}(\emptyset(Q)(\emptyset(K))^TV)\hat{D}^{-1} = \mathrm{diag}(\emptyset(Q)(\emptyset(K))^T \mathbf{1_{K}}) \]
其中 \(\emptyset(\cdot)\) 是一个核函数,用于逼近原始注意力方程中的 \(A\) 矩阵。
解码器的输出是 \(X^{Out}\),其中
\[ X^{Out} = \mathrm{Performer}(X^{\mathrm{Dec-Input}}) \in \mathbb{R}^{19.266\times f} \]
为了预测表达值,删除了 \(T\) 和 \(S\) 的嵌入,并遵循 MLP 将 \(X^{Out}\) 投影到标量。这些标量形成了预测向量 \(P\),其中
所有参数 \(\Theta = \{ \mathbf{E}_{i},\mathbf{T}_{i}^G,\Theta_{\mathrm{Encoder}},\Theta_{\mathrm{Decoder}},\Theta_{\mathrm{MLP}} \}\) 在预训练期间进行了优化。不同模型的详细超参数设置可以在补充表 7 中找到。
RDA pretraining task
scFoundation 使用 RDA 基因表达预测任务训练模型。对于每个原始预训练单细胞基因表达向量,使用分层贝叶斯下采样策略来生成其低总计数变体或未更改的配置文件作为输入向量。对原始和输入基因表达进行归一化和对数转换,并将原始和输入向量的总计数分别设置为两个总计数指标 T 和 S。基因表达标准化后,去除细胞原始总计数值。通过令牌重新引入这些信息,相信它可以增强模型的预训练性能,因为单元格中的丢失通常与总计数值相关。抽样策略及计数指标计算详情请参阅附注 9 及附注 10。
然后随机屏蔽输入向量的基因表达。在本研究中,使用 30% 作为零值和非零值的掩蔽比。然后,将屏蔽输入向量与两个总计数指标 T 和 S 连接起来,并输入到模型中。在获得模型预测的原始基因表达后,对预测值和原始值之间的掩蔽基因进行回归损失(补充说明 11)。如果输入向量不变,模型就会学会捕获单个细胞内基因之间的关系。如果输入向量是低总计数变体,则模型会了解具有不同读取深度的细胞之间的关系。采取下采样策略(补充表 8)和回归损失(补充图 15)的消融研究(补充说明 1)表明,当前设置可以促进学习细胞特征。
scFoundation 的整体模型架构如补充图 16 所示。有关模型和预训练实现,请参阅补充说明 12。
Read-depth enhancement analysis
对于基因表达预测评估,从 5000 万个单细胞数据中采样了 10,000 个总计数较高(高于 1,000)的细胞作为验证数据集。这 10,000 个细胞在训练阶段被排除。然后,使用二项式分布来生成低总计数基因表达向量并将其输入到 scFoundation 模型中。考虑到 0 表达值在下采样后不会发生变化,仅评估非零基因表达值。除了使用 MSE 作为评价指标外,还使用了 MRE,它可以反映相对误差
\[ \mathrm{MRE} = \frac{1}{|M|} \sum_{i=0}^{|M|} \frac{(X_{i}-P_{i})^2}{X_{i}}. \]
为了进行聚类分析,从 scFoundation 和 scVI 编码器获得了细胞嵌入。对于其他人, scFoundation 得到了估算的基因表达谱。所有方法均使用默认参数设置。然后,按照 SCANPY pbmc3k 教程并通过函数“sc.tl.leiden”获取细胞簇。
对于聚类结果的评估, scFoundation 首先使用 ARI 和 NMI(scikit-learn 包)作为指标来评估不同方法得到的聚类结果与实际细胞类型标签的一致性程度。考虑到聚类标签的获取也会受到聚类算法选择的影响, scFoundation 使用 SIL 作为另一个评价指标,它衡量真实细胞类型标签在不同方法给出的细胞邻域图上的聚集程度,从而,与聚类算法的选择无关,反映了单元表示的内在属性。