2024 单细胞 RNA
测序数据应用系列 1 预处理
对于单细胞 RNA
测序数据我们通常会进行一个预处理过程,这个也和大多数机器学习和深度学习项目一致。这里我们介绍以
h5 文件格式存储的单细胞 RNA
测序数据的预处理。预处理主要是对表达矩阵进行归一化和去除低表达基因和细胞。
数据预处理
读取计数数据(count data)
以 “ZINB-Based Graph Embedding Autoencoder for Single-Cell RNA-Seq
Interpretations” (scTAG)中读取存储单细胞 RNA 测序数据为例。
数据格式介绍
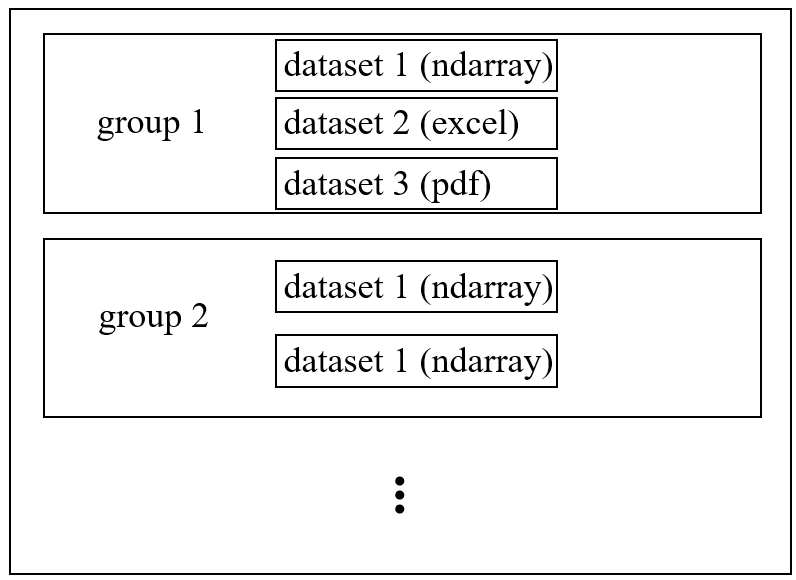
首先对 scTAG 中 h5 文件进行简要介绍。H5 文件是层次数据格式第 5
代的版本(Hierarchical Data
Format,HDF5),它是用于存储科学数据的一种文件格式和库文件。
h5 文件中有两个主要结构:组“group”和数据集“dataset”。一个 h5 文件就是
“group” 和 “dataset” 二合一的容器,group 名称为key,datasets 为
value。
代码实现
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
| import numpy as np
def prepro(filename):
"""
函数用于预处理数据文件。
参数:
filename (str): 数据文件的路径名。
返回值:
tuple: 一个包含两个元素的元组:
- X (numpy.ndarray): 预处理后的表达矩阵。
- cell_label (numpy.ndarray): 细胞类型的标签数组。
"""
data_path = filename
mat, obs, var, uns = read_data(data_path, sparsify=False, skip_exprs=False)
if isinstance(mat, np.ndarray):
X = np.array(mat)
else:
X = np.array(mat.toarray())
cell_name = np.array(obs["cell_type1"])
cell_type, cell_label = np.unique(cell_name, return_inverse=True)
return X, cell_label
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
| import h5py
import scipy as sp
import numpy as np
import scanpy as sc
import pandas as pd
def read_data(filename, sparsify=False, skip_exprs=False):
"""
从HDF5格式的数据文件中读取数据。
参数:
filename (str): HDF5文件的路径名。
sparsify (bool): 是否将表达矩阵转换为稀疏矩阵,默认为False。
skip_exprs (bool): 是否跳过表达矩阵的加载,默认为False。
返回值:
tuple: 包含四个元素的元组:
- mat (scipy.sparse.csr_matrix 或 numpy.ndarray): 表达矩阵。
- obs (pandas.DataFrame): 观测信息。
- var (pandas.DataFrame): 变量信息。
- uns (dict): 未分类信息。
"""
with h5py.File(filename, "r") as f:
obs = pd.DataFrame(dict_from_group(f["obs"]), index=utils.decode(f["obs_names"][...]))
var = pd.DataFrame(dict_from_group(f["var"]), index=utils.decode(f["var_names"][...]))
uns = dict_from_group(f["uns"])
if not skip_exprs:
exprs_handle = f["exprs"]
if isinstance(exprs_handle, h5py.Group):
mat = sp.sparse.csr_matrix((
exprs_handle["data"][...],
exprs_handle["indices"][...],
exprs_handle["indptr"][...]), shape=exprs_handle["shape"][...])
else:
mat = exprs_handle[...].astype(np.float32)
if sparsify:
mat = sp.sparse.csr_matrix(mat)
else:
mat = sp.sparse.csr_matrix((obs.shape[0], var.shape[0]))
return mat, obs, var, uns
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
| import h5py
class dotdict(dict):
"""
创建一个字典的子类,它允许使用点符号(.)来访问字典键,就像访问对象属性一样。
方法:
__getattr__(self, key): 通过点符号访问字典中的值。
__setattr__(self, key, value): 通过点符号设置字典中的值。
__delattr__(self, key): 通过点符号删除字典中的值。
"""
__getattr__ = dict.get
__setattr__ = dict.__setitem__
__delattr__ = dict.__delitem__
def read_clean(data):
"""
清理从HDF5文件读取的数据。
参数:
data (numpy.ndarray): 需要清理的数据数组。
返回:
数据清理后的结果,可能是单个值或解码后的字符串数组。
断言:
确保传入的数据是NumPy数组类型。
"""
assert isinstance(data, np.ndarray)
if data.dtype.type is np.bytes_:
data = utils.decode(data)
if data.size == 1:
data = data.flat[0]
return data
def dict_from_group(group):
"""
递归地将HDF5 Group对象转换为Python字典。
参数:
group (h5py.Group): HDF5 Group对象,包含数据集和其他Group。
返回:
dotdict: 包含所有子组和数据集的字典,可以使用点符号访问。
"""
assert isinstance(group, h5py.Group)
d = dotdict()
for key in group:
if isinstance(group[key], h5py.Group):
value = dict_from_group(group[key])
else:
value = read_clean(group[key][...])
d[key] = value
return d
|
预处理(Pre-processing)
原始 scRAN-seq 读取计数数据由 Python 包 SCANPY
进行预处理。首先,过滤掉任何细胞中没有计数的基因。其次,计算大小因子,并根据文库大小对读取计数进行归一化,因此细胞间的总计数相同。形式上,如果我们将细胞
\(i\) 的库大小(总读取计数数)表示为
\(s_i\),则细胞 \(i\) 的大小因子为 \(s_i/median(s)\)。最后一步是对读数进行对数变换和缩放,以便计数值遵循单位方差和零均值。预处理的读取计数矩阵为模型的的输入。
代码实现
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
| import scipy as sp
import scanpy as sc
def normalize(adata, copy=True, highly_genes=None, filter_min_counts=True, size_factors=True, normalize_input=True, logtrans_input=True):
"""
对单细胞RNA测序数据进行标准化预处理。
参数:
adata (AnnData 或 str): 输入的AnnData对象或文件路径。
copy (bool): 是否在操作前复制输入数据,默认为True。
highly_genes (int 或 None): 选择高变基因的数量。如果为None,则不选择。
filter_min_counts (bool): 是否过滤掉表达量低的基因和细胞,默认为True。
size_factors (bool): 是否计算并应用大小因子(size factors)进行标准化,默认为True。
normalize_input (bool): 是否对输入数据进行标准化,默认为True。
logtrans_input (bool): 是否对输入数据进行对数转换,默认为True。
返回:
AnnData: 经过预处理后的AnnData对象。
"""
if isinstance(adata, sc.AnnData):
if copy:
adata = adata.copy()
elif isinstance(adata, str):
adata = sc.read(adata)
else:
raise NotImplementedError
norm_error = 'Make sure that the dataset (adata.X) contains unnormalized count data.'
assert 'n_count' not in adata.obs, norm_error
if adata.X.size < 50e6:
if sp.sparse.issparse(adata.X):
assert (adata.X.astype(int) != adata.X).nnz == 0, norm_error
else:
assert np.all(adata.X.astype(int) == adata.X), norm_error
if filter_min_counts:
sc.pp.filter_genes(adata, min_counts=1)
sc.pp.filter_cells(adata, min_counts=1)
if size_factors or normalize_input or logtrans_input:
adata.raw = adata.copy()
else:
adata.raw = adata
if size_factors:
sc.pp.normalize_per_cell(adata)
adata.obs['size_factors'] = adata.obs.n_counts / np.median(adata.obs.n_counts)
else:
adata.obs['size_factors'] = 1.0
if logtrans_input:
sc.pp.log1p(adata)
if highly_genes != None:
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5,
n_top_genes=highly_genes, subset=True)
if normalize_input:
sc.pp.scale(adata)
return adata
|
参考
[1] H5文件简介以及python对h5文件的操作
[2] .h5文件的写入和读取(HDF5)
[3] .h5图像文件(数据集)的读取并存储工具贴(二)
[4] ZINB-based Graph
Embedding Autoencoder for Single-cell RNA-seq Interpretations